order histories, retained contact details for faster checkout, review submissions, and special promotions.
Forgot password?
order histories, retained contact details for faster checkout, review submissions, and special promotions.
Location
Corporate Headquarters
Vector Laboratories, Inc.
6737 Mowry Ave
Newark, CA 94560
United States
Telephone Numbers
Customer Service: (800) 227-6666 / (650) 697-3600
Contact Us
Additional Contact Details
order histories, retained contact details for faster checkout, review submissions, and special promotions.
Forgot password?
order histories, retained contact details for faster checkout, review submissions, and special promotions.
Alzheimer’s Disease
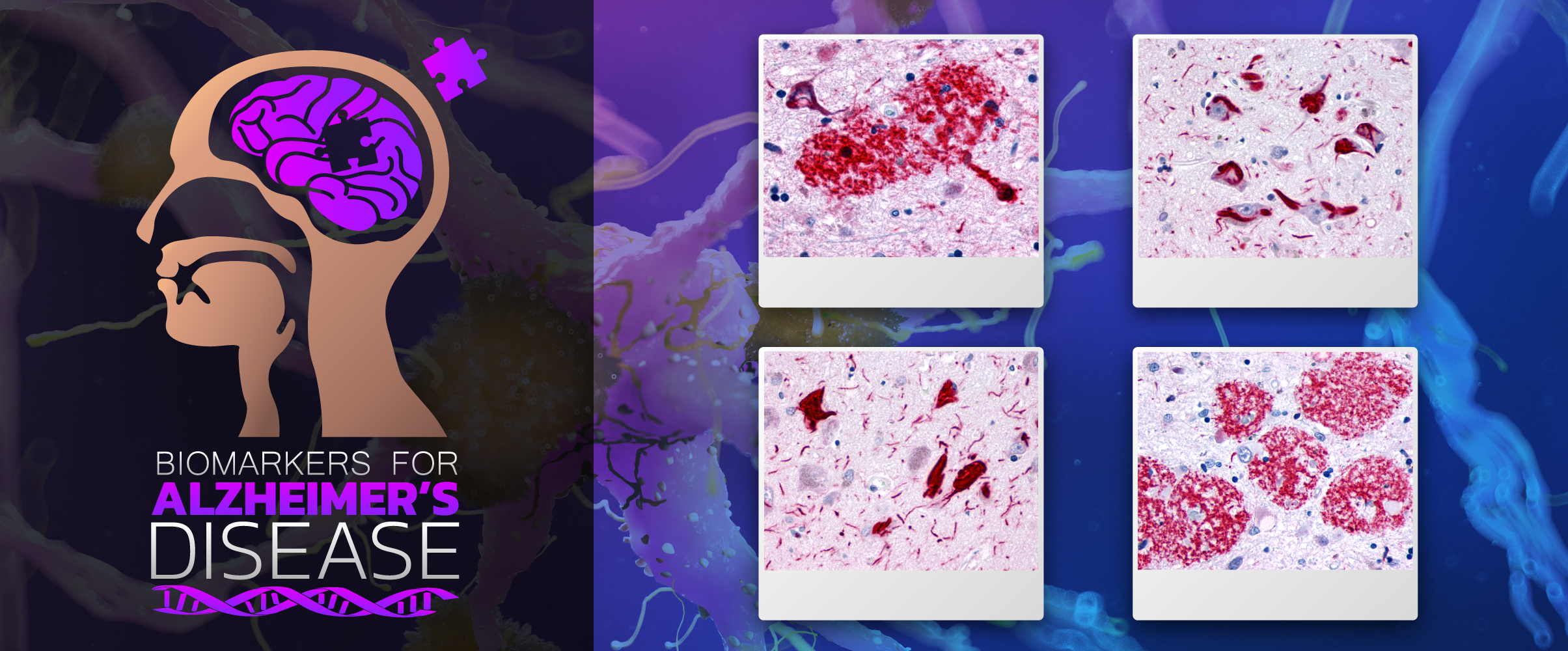
Alzheimer’s Disease (AD) is the most common progressive form of dementia. It is typically late onset (95% age >60 years), characterized by neuroimaging findings of cerebral cortical atrophy with cerebral amyloid angiopathy and CSF fluid measurements of amyloid-beta peptide, total or phosphorylated tau (Cohen Ad 2018). Neuropathological findings that are diagnostic include neuritic plaques containing beta-amyloid, neurofibrillary tangles consisting of hyper-phosphorylated microtubule associated protein tau, and amyloid angiopathy, described by accumulations of amyloid-beta proteins in the vascular smooth muscle of small cerebral arteries and capillaries.
Genetics
Approximately 25% of AD cases are familial, and multiple susceptibility genes have been identified that increase the risk for disease development. The gene APOE has three allelic variants (e2, e3, and e4) that encode different isoforms of the protein, and the presence of the APOE e4 allele in either the heterozygous (APO e3/e4) or homozygous state (APO e4/e4) increases the risk for early and late onset AD. Other genes that have been associated with early-onset familial AD include APP, PSEN1 and PSEN2. The dementia phenotype is similar to late-onset AD, but the age of onset is usually in the 40s and 50s. Approximately 20 other genes have also been identified that increase the risk for developing AD, including ABCA7, AKAP9, BIN1, CASS4, CD2AP, CD33, CLU, EPHA1, FERMT2, HLA-DRB5/DRB1, INPP5D, MEF2C, MS4A6A/MS4A4E, PICALM, PLD3, PTK2B, SORL1, TREM2, and UNC5C.
Biomarkers
Despite these association with specific clinical and genetic markers, the identification of sensitive and specific, predictive blood-based biomarkers for AD has been challenging (Khoury R 2019; Zetterberg 2019; Liu Shi 2018). So far, biomarkers have fallen into three categories; beta-amyloid A-beta deposits, hyper-phosphorylated tau aggregates, and markers for neural degeneration and axonal injury, such as plasma neurofilament light (NfL). In addition, inflammatory markers, as well as proteins involved in the complement cascade have shown association with cognitive decline and performance (Zetterberg 2019). Blood markers such as APOE that are associated with neocortical amyloid beta deposition (as measured by PET scans) have been studied, and others have correlated pancreatic polypeptide and IgM with neocortical amyloid beta deposition (Kiddle, 2012; Zetterberg 2019). The search for novel biomarkers has brought to light a number of proteins that show correlations with amyloid and tau pathways, inflammation, the complement cascade, and various hormones. Clusterin, complement C3, gamma fibrinogen, ficolin-1, APOE, PPY, alpha- 2-macroglobulin, alpha-1-antitrypsin, and pancreatic polypeptide are the most commonly reported AD biomarkers in the literature (reviewed by Shi 2018).
Pathogenesis
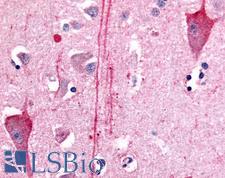
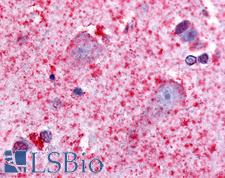
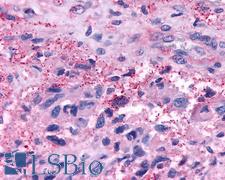
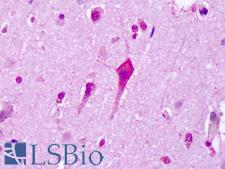
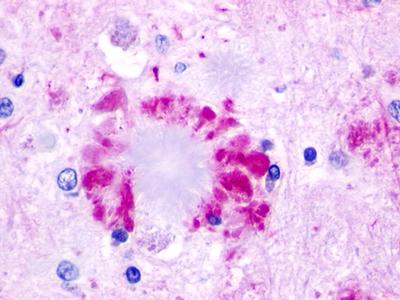
The pathogenesis of AD has been attributed to the extracellular accumulation of aggregates of amyloid beta (Aβ) and intracellular hyperphosphorylated microtubule-associated 𝜏 (tau) proteins in cortical and limbic neurons within the brain (Tiwari 2019). Aβ plaques initially develop in the basal, temporal, and orbitofrontal neocortex and in later stages progress throughout the neocortex, hippocampus, amygdala, diencephalon, and basal ganglia. Late stage AD can also show plaques in the mesencephalon, lower brain stem, and cerebellum. Tau-tangles form in the locus coeruleus and entorhinal cortex and can spread to the hippocampus and neocortex.
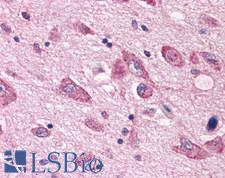
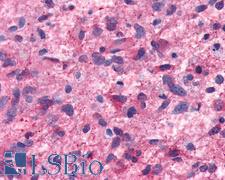
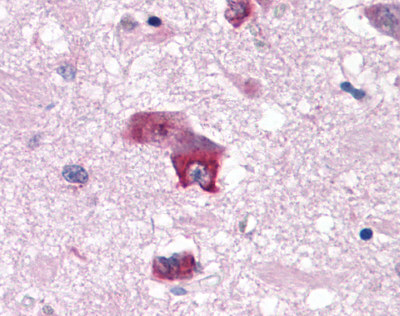
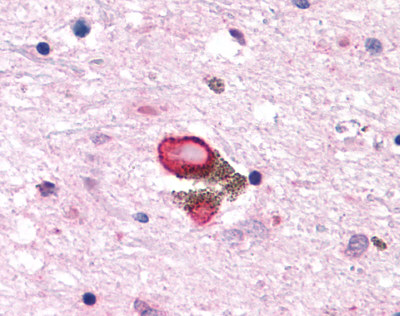
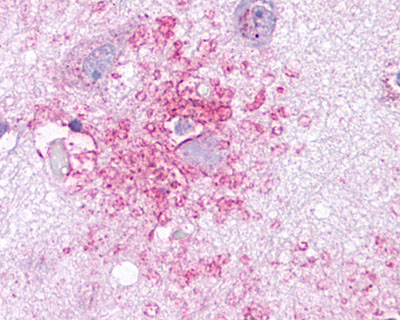
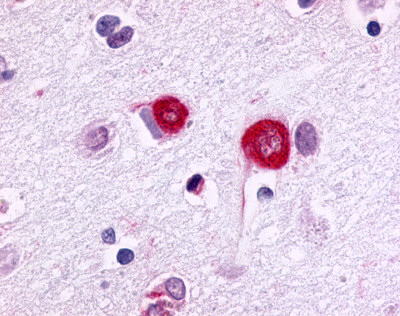
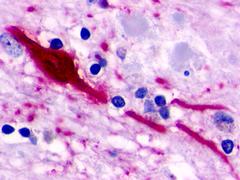
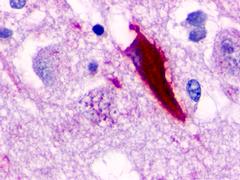
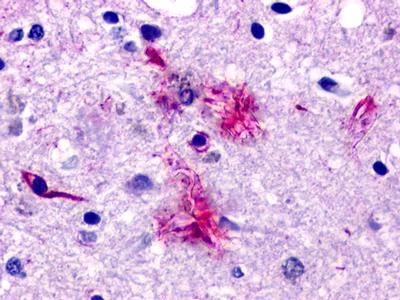
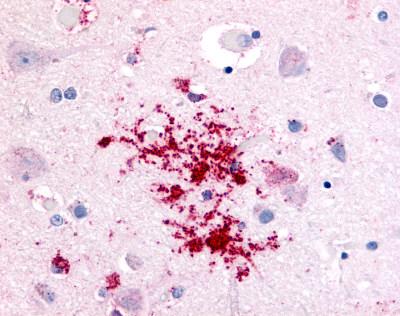
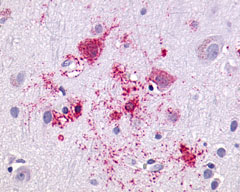
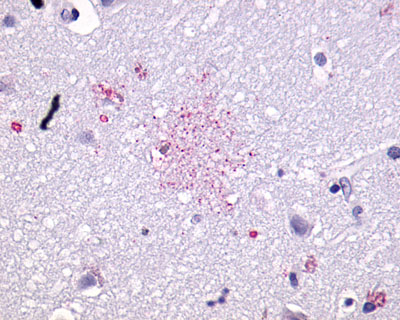
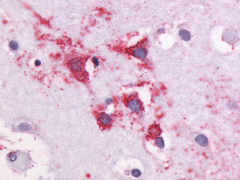
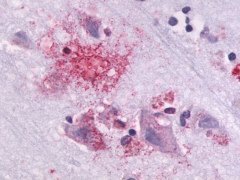
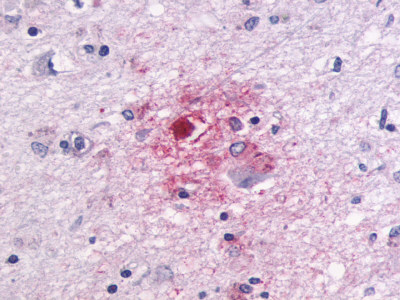
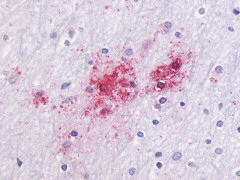
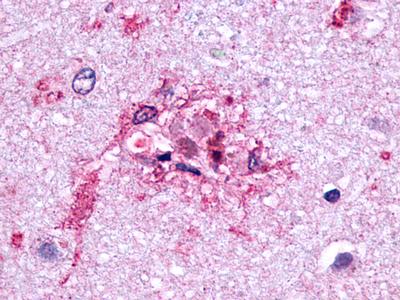
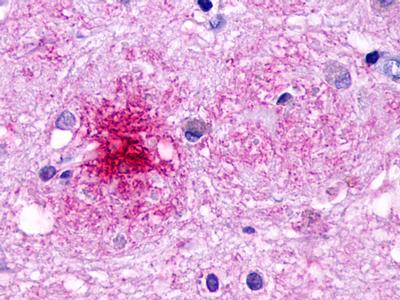
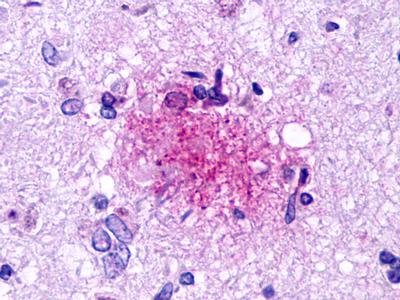
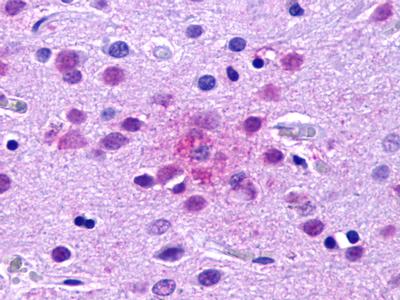
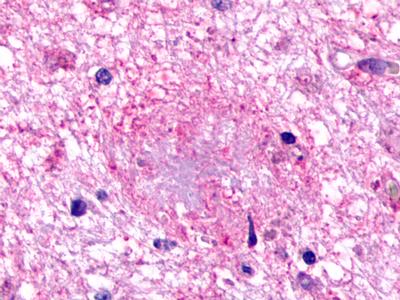
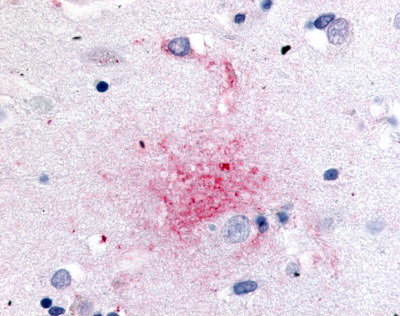
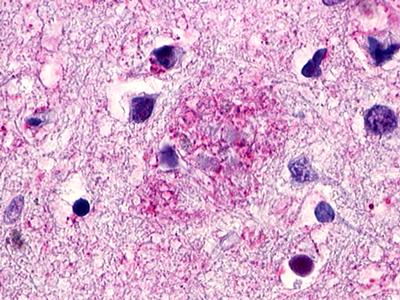
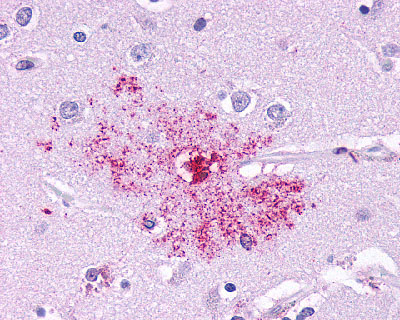
The neuritic plaques found in the brains of Alzheimer’s patients are extracellular foci of amyloid beta protein deposition associated with axonal and dendritic injury, and are accompanied by microglia expressing CD45 and HLA-DR, surrounded by reactive astrocytes. Many of these plaques are found in the limbic and association cortices, but can also be found in the thalamus, caudate, putamen, and cerebellum.
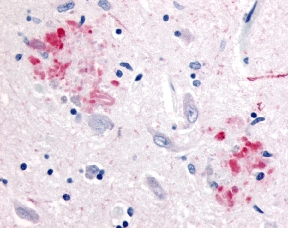
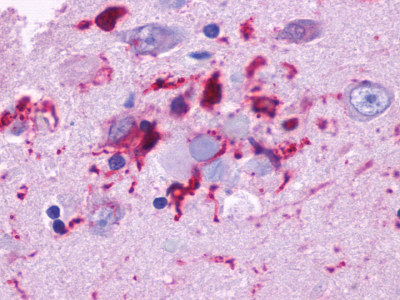
Amyloid accumulations occur from the altered cleavage of amyloid precursor protein (APP) by β and γ secretases (BACE1) to produce insoluble Aβ fibrils. These fibrils oligomerize, enter synaptic clefts, and interfere with synaptic signaling, as well as polymerize into plaques. The polymerization activates kinases that lead to hyperphosphorylation of tau protein and its polymerization into neurofibrillary tangles. The accumulation of plaques and tangles triggers microglial activation and a local inflammatory response (Crews L 2010).
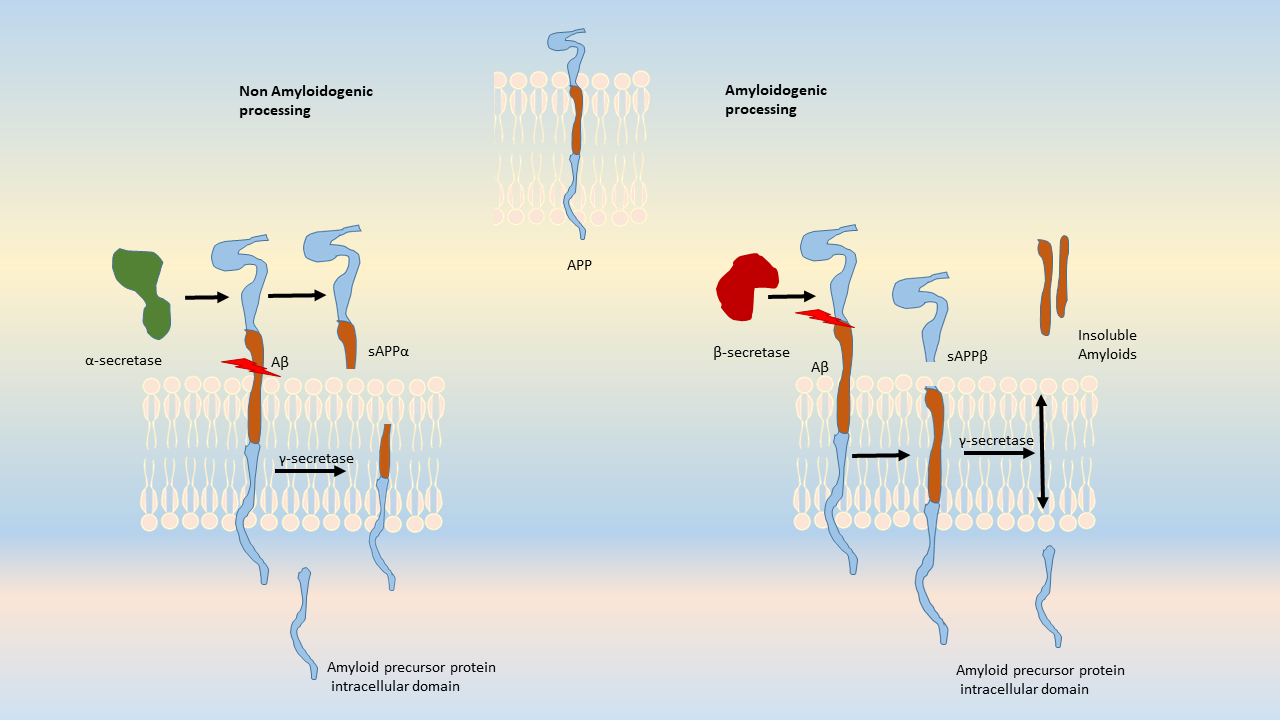
Amyloid precursor protein is an integral transmembrane protein that moderates cell growth and motility, including neurite outgrowth, by releasing soluble ectodomains upon cleavage of APP. The enzymes responsible for normal cleavage include α-secretases such as ADAM10 and ADAM17 metalloproteases, and γ-secretases. In diseased states, APP generates amyloidogenic fragments by an altered cleavage process that results in insoluble Aβ peptides. Aβ is released from APP after cleavage by BACE-1, γ-secretase, and a protease that is comprised of four proteins: presenilin, nicastrin, Aph1 and Psen2. This complex contributes to the production of insoluble and neurotoxic Aβ fragments that polymerize into plaques (reviewed by Tiwari, 2019). These Aβ polymers aggregate to block ion channels, increase oxidative stress, and diminish energy metabolism, resulting in neuronal cell death.
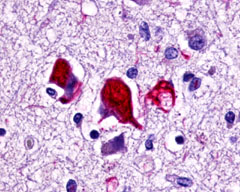
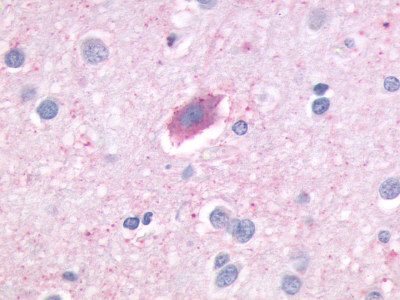
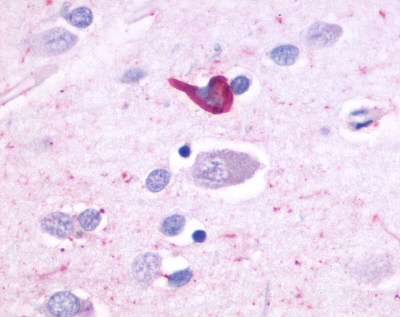
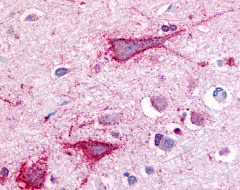
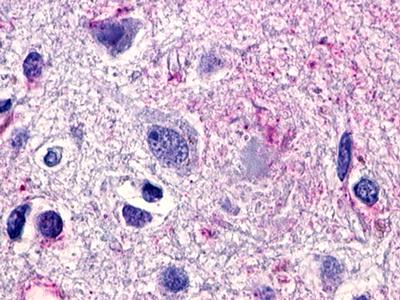
Neurofibrillary tangles are large, non-membrane-bound bundles of abnormal fibers that are found in the cytoplasm of neurons in the frontal, temporal, parietal, entorhinal and occipital cortex, hippocampus, parahippocampus, and amygdala in patients with AD.
Neurofibrillary tangles (NFTs) are the result of hyperphosphorylation of the microtubule-associated 𝜏-protein (tau). The 𝜏 protein co-assembles with tubulin to form matured microtubules. When Aβ accumulates in the cell microenvironment, the released kinases hyperphosphorylate the 𝜏-protein, causing it to oligomerize and aggregate into NFTs. These highly insoluble accumulations of NFTs within the neuronal cytoplasm result in a loss of communication between neurons and ultimately apoptosis. Other kinases are also involved in the phosphorylation of 𝜏 protein or processing of APP, including GSK3β, CDK5, Protein Kinase C, Protein Kinase A, ERK2, caspase 3, and caspase 9. In addition, other kinases implicated in hyperphosphorylation of 𝜏 protein include MAPK, ERK1, MEK, MARK, JNKs, p38, and PKA.
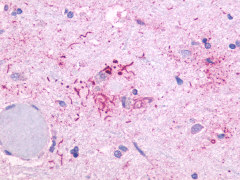
Microglial infiltration during plaque formation has also been shown to exacerbate AD pathogenesis. Extracellular and intracellular Aβ plaques and NFTs cause toxicity and microglial infiltration and the release of pro-inflammatory cytokines and chemokines, triggering immune responses in plaque areas. Microglia bind to Aβ via cell surface receptors such as SCARA1, CD36, CD14, alpha-6 beta-1 integrin, CD47, and Toll-like receptors. Microglial clearance of Aβ can also be compromised in patients with TREM2 mutations, a cell surface receptor that is required for phagocytic clearance of neuronal debris.
Other genes that are associated with AD include the presenilins (PSEN1 and PSEN2), which belong to the γ-secretase family, and whose mutations are associated with early onset AD. Mutant forms of PSEN1 and PSEN2, both part of the four proteins comprising the g-secretase complex (along with nicastrin and Aph1), have been shown to produce more toxic forms of amyloid, which contribute to disease progression.
G Protein-Coupled Receptors
In addition to these markers, various families of G protein-coupled receptors (GPCRs) have been identified to have putative roles in CNS disorders as well as Alzheimer’s disease (Dal Pra, I. 2019; Alavi 2018; Xu, 2020, Zhao, J 2016). The vertebrate GPCRs are a superfamily of membrane proteins comprising five distinct families on the basis of structural similarity and sequence, including rhodopsin (family A), secretin (family B), glutamate (family C), adhesion, and Frizzled/Taste2. These families all share a structure which consists of seven transmembrane helices connected to three extracellular and three intracellular loops. Despite a common structure, they have distinct roles in signal-transduction and regulatory processes. GPCRs have been implicated in the pathogenesis of Alzheimer’s disease and have been shown to bind to BACE1 and gamma secretase, both involved in the hydrolytic processing of APP (Zhao, 2016).
GPCRs implicated in AD include well-described GPCRS such as the gamma-aminobutyric acid B receptors, muscarinic cholinergic receptors (M1-3 AChR), the metabotropic glutamate receptors mGluR1-8, histamine receptors, delta opioid receptor, chemokine receptors, and calcium-sensing receptor CaSR, as well as orphan receptors such as GPR3, GPR6, GPR17, GPR26, GPR37, GPR39, GPR40, GPR50, GPR52, GPR54, GPR55, GPR68, GPR85, GPR88, GPR103, and GPR139 (Dal Pra 2019; Alavi, 2018; Xu 2020, Table 1). Other groups of GPCRs have also been identified that appear to be primarily involved in the microglial activation response to AD (Haque 2018). The GPCRs are variously involved in neurodevelopment, cytoskeletal organization, cell migration and synapse development, synaptic plasticity, endocytosis and phagocytosis, microglial or astroglial function, or immune regulation, so it is unsurprising that a number of these genes may be involved in the pathological progression of AD (see Table 1, GPCRs Involved in AD).
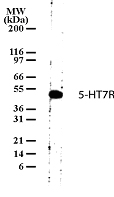
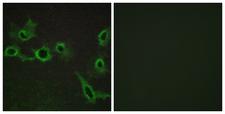
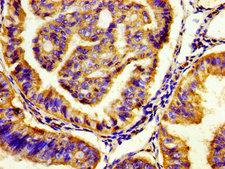
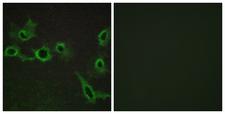
LifeSpan has also generated and tested antibodies to over 350 GPCRs by IHC on brain samples from patients with Alzheimer’s disease. Positive staining of senile plaques, neurofibrillary tangles, or overexpression in astrocytes in areas of injury was observed in IHC using antibodies to 33 different GPCRs (Table 1), most of which have been associated with AD progression or as a marker in previous (non-IHC) studies, demonstrating the widespread involvement of this class of proteins in neurodegenerative disorders such as AD.
Download the full GPCR table here
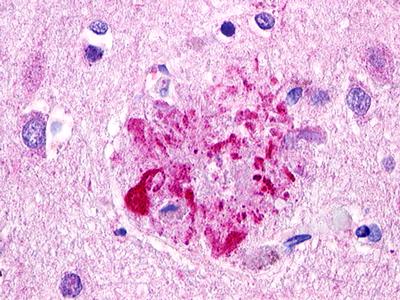
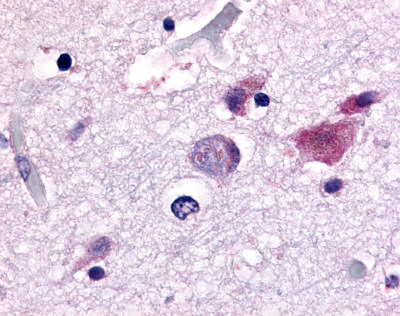
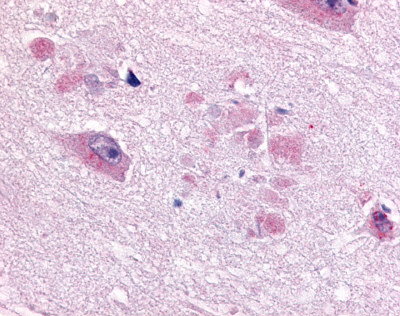
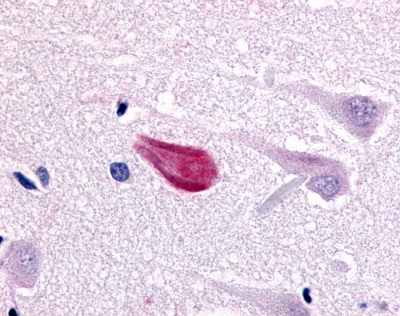
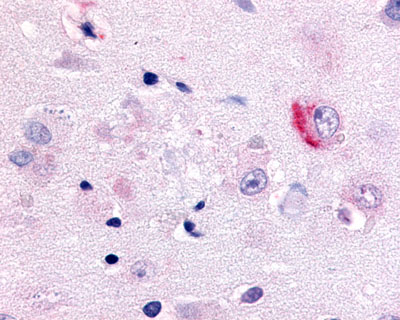
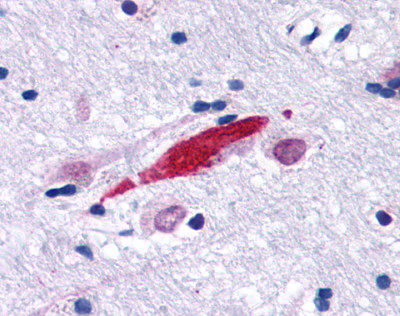
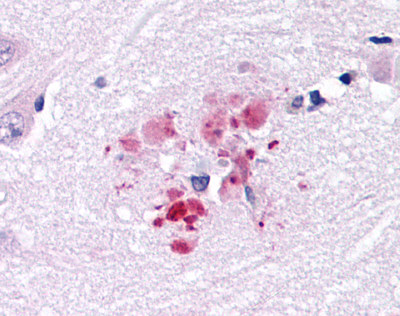
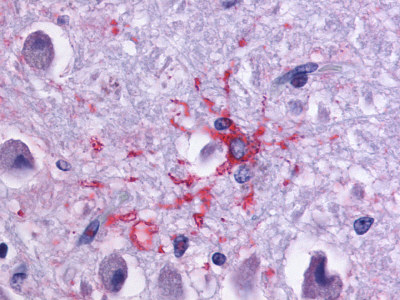
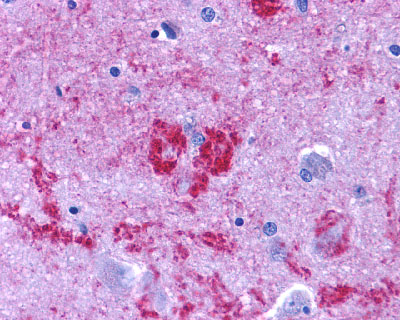
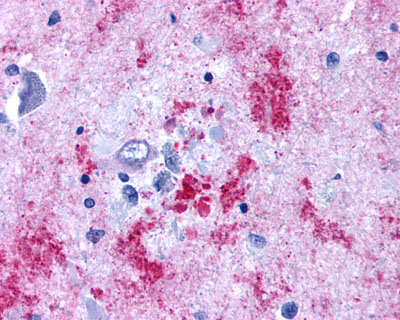
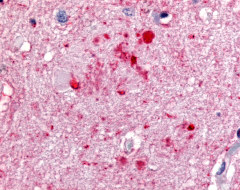
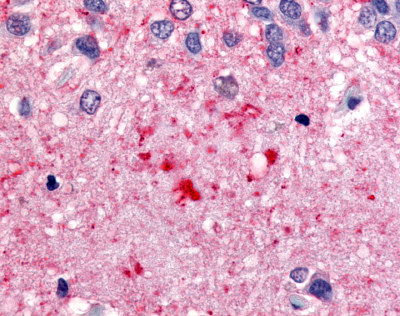
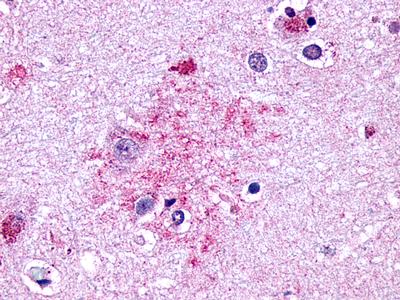
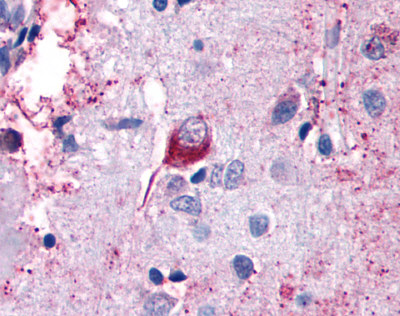
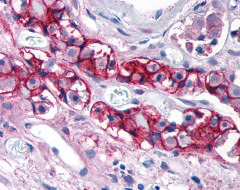
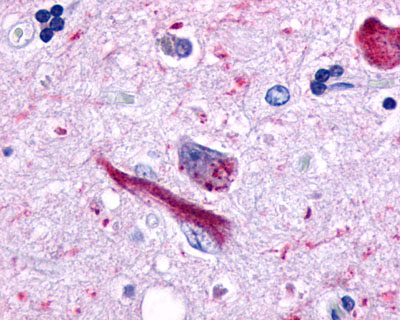
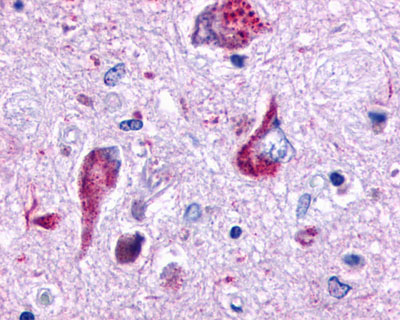
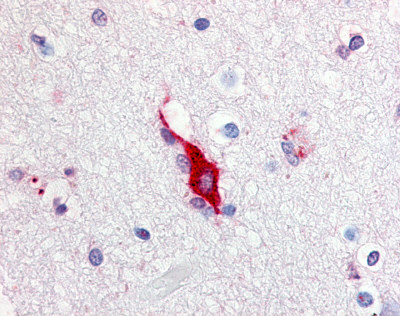
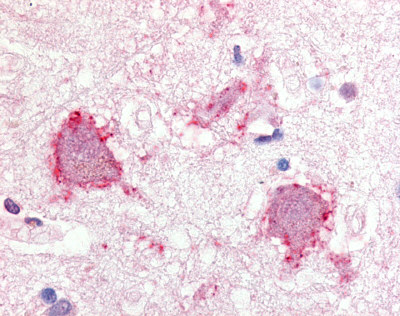
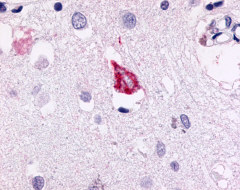
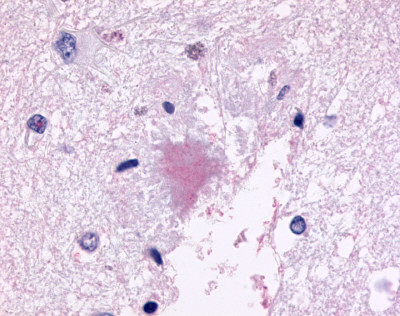
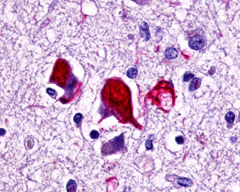
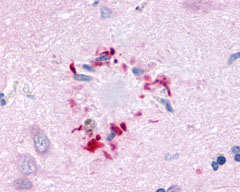
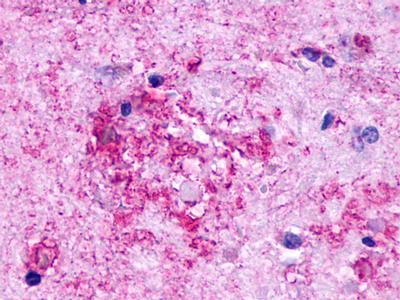
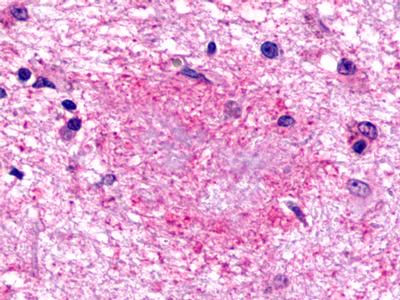
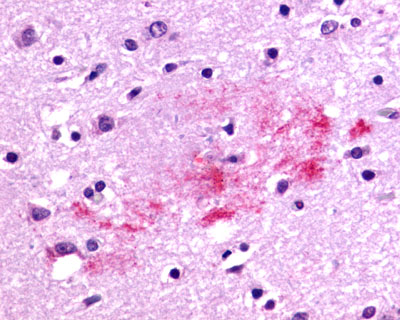
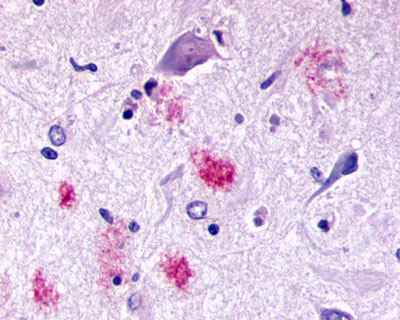
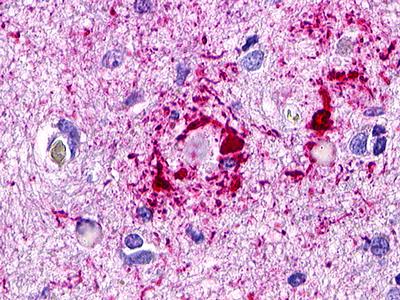
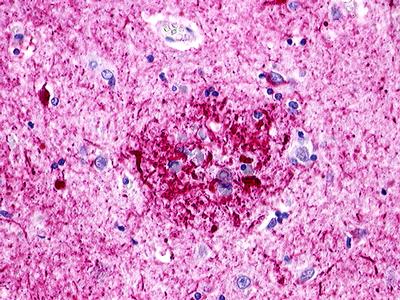
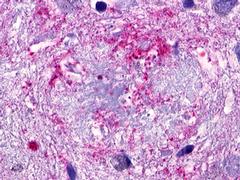
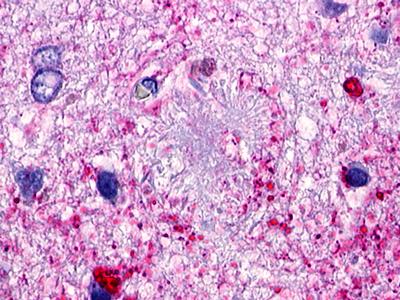
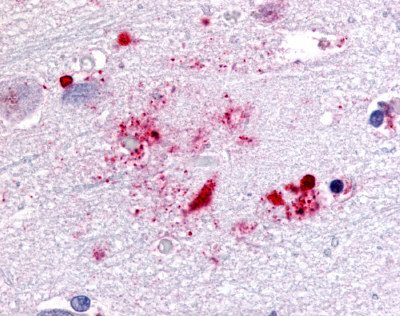
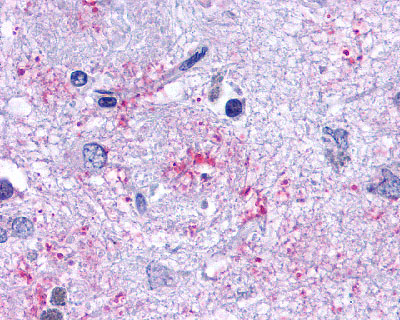
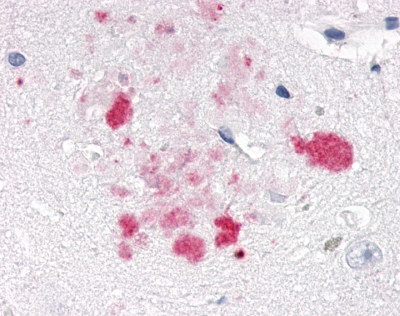
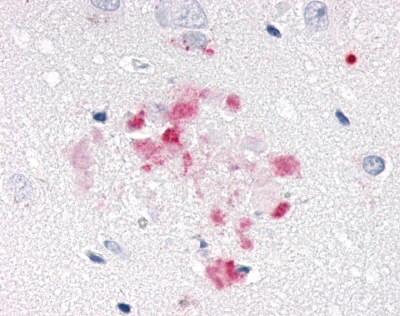
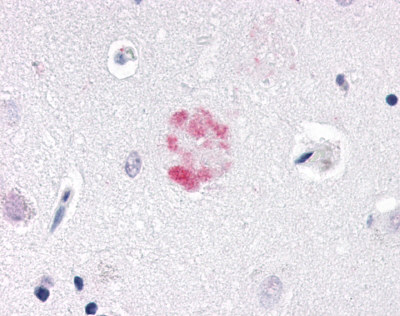
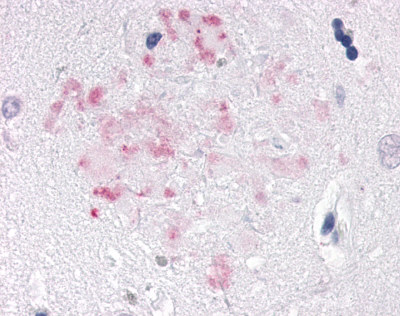
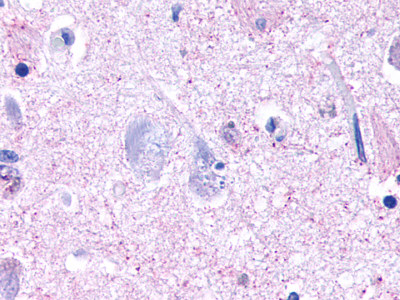
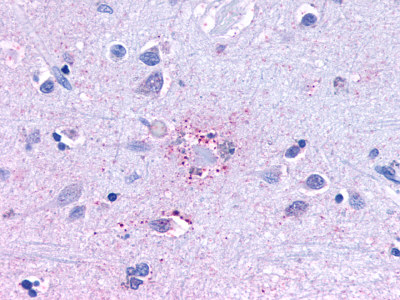
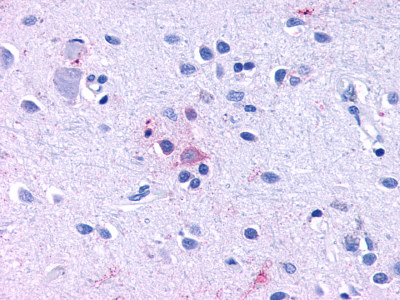
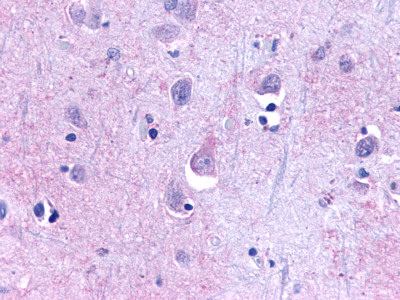
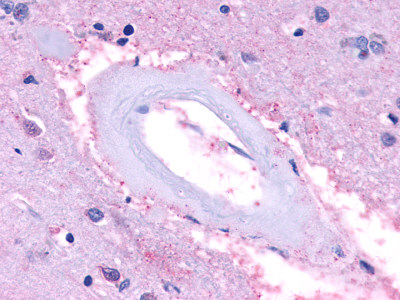
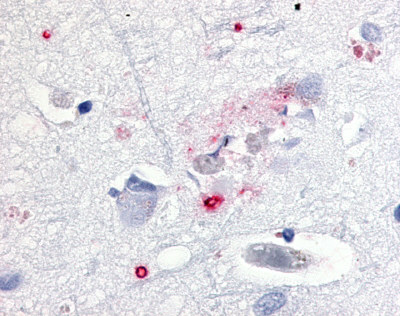
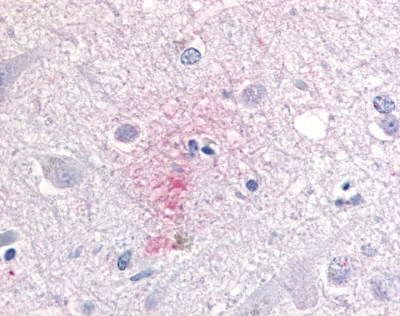
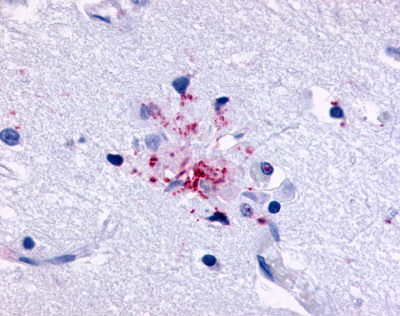
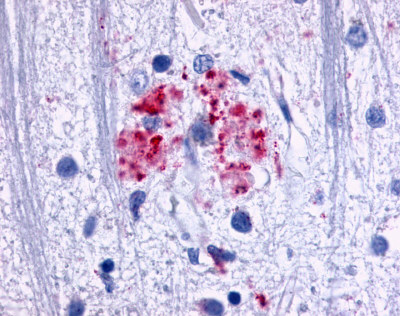
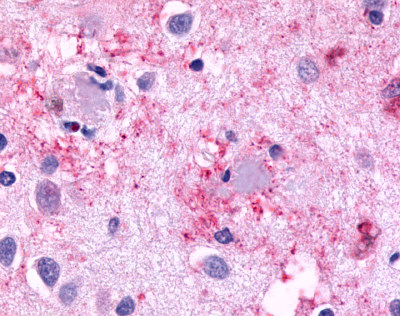
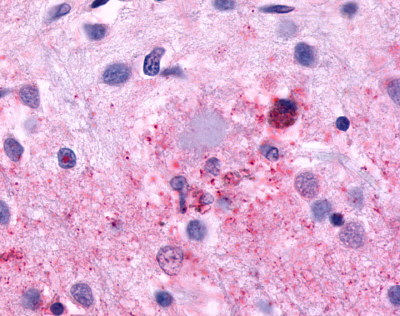
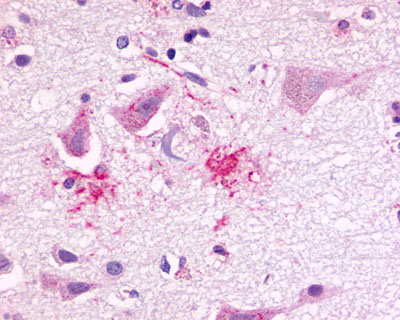
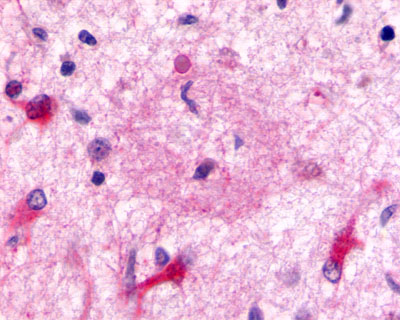
Therapeutics
Although there are no current drugs that can reverse the progressive dementia of AD, a number of therapeutic agents are used to counter the neurotransmitter shortage and imbalance that occurs in this disease. These include acetylcholinesterase inhibitors (AChEIs) such as donepezil, galantamine, and rivastigmine that increase the availability of acetylcholine at synapses. NMDA receptor antagonists such as memantine are also used to block NMDA-mediated ion flux and reduce the elevated glutamate levels that can be toxic to neurons. Other drugs in clinical trials target the accumulation of abnormal proteins (amyloid and tau) within the extracellular spaces and neurons, to prevent their misfolding, or removing the toxic aggregates and reducing the plaque burden. (Yiannopoulou, 2020). These include γ-secretase inhibitors, BACE inhibitors, α-secretase modulators, amyloid aggregation inhibitors, and immunotherapies designed to promote Aβ clearance. Kinase inhibitors are also under study as therapeutic targets to prevent the hyperphosphorylation or aggregation of tau protein. Other therapeutic approached being tested in clinical trials include those that target neuroprotection and reduce neuronal hyperactivity, anti-inflammatory agents and complement blockers, growth factors to promote neuronal regeneration, and stem cell therapies.
References
- Alavi, MS et al. (2018) Orphan G protein-coupled receptors: The role in CNS disorders. Biomedicine & Pharmacotherapy. 98:222-232. https://doi.org/10.1016/j.biopha.2017.12.056
- Bird, TD. (2018) Alzheimer Disease Overview. NCBI Gene Reviews. https://www.ncbi.nlm.nih.gov/books/NBK1161/
- Cohen, AD et al. Fluid and PET biomarkers for amyloid pathology in Alzheimer’s disease. (2019). Mol Cell Neurosci. 97:3-17.https://doi:10.1016/j.mcn.2018.12.004. Epub 2018 Dec 8..
- Crews, L Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. (2010). Hum Mol Genet. 19(R1):R12-R20. https://doi:10.1093/hmg/ddq160
- Dal Pra, I et al. (2019). Family C G-Protein-Coupled Receptors in Alzheimer’s Disease and Therapeutic Implications. Front. Pharmacol. 28 Oct. https://doi.org/10.3389/fphar.2019.01282
- Kiddle, SJ et al. Plasma based markers of [11C] PiB-PET Brain Amyloid Burden. (2012). PloS One Sept 24 https://doi.org/10.1371/journal.pone.0044260
- Khoury, R and Ghossoub, E. Diagnostic biomarkers of Alzheimer’s disease: a state-of-the-art review. (2019) Biomarkers in Neuropsychiatry Vol1:100005. https://doi.org/10.1016/j.bionps.2019.100005
- Liu, S et al. A Decade of Blood Biomarkers for Alzheimer’s Disease Research: An Evolving Field, Improving Study Designs, and the Challenge of Replication. (2018). J. Alzheimer’s Disease 62(3):1181-1198
- Rychlik, M et al. (2020) Zinc-mediated Neurotransmission in Alzheimer’s Disease: A potential role of the GPR39 in Dementia. Curr Neuropharmacol. 18(1):2-13. https://doi: 10.2174/1570159X17666190704153807.
- Selkoe, DJ. (2001) Alzheimer’s Disease: Genes, Proteins, and Therapy. Physiological Reviews. 81 (2): 741-766. https://doi:10.1152/physrev.2001.81.2.741.
- Thathiah, A. et al. (2011). The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nature Reviews Neuroscience. 12:73-87. https://doi:10.1038/nrn2977
- Tiwari S et al. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. (2019) Int J Nanomedicine. 14:5541-5554. https://doi.org:10.2147/IJN.S200490
- Xu, Y et al. (2020). GPR68 deletion impairs hippocampal long-term potentiation and passive avoidance behavior. Molecular Brain. 13:132. https://doi.org/10.1186/s13041-020-00672-8
- Yiannopoulou, KG and Papageorgiou SG. Current and Future Treatments in Alzheimer Disease: An Update. (2020) J Cent Nerv Syst Dis 12:1179573520907397. https://doi.org:10.1177/1179573520907397
- Zetterbert, H and Burnhan, SC. Blood-based molecular biomarkers for Alzheimer’s disease. (2019) Molecular Brain 12 (26). https://doi.org/10.1186/s13041-019-0448-1
- Zhao, J et al. (2016). G Protein-Coupled Receptors (GPCRs) in Alzheimer’s Disease: A Focus on BACE1 Related GPCRs. Frontiers in Aging Neuroscience Vol8, Article 58:1-15. https://doi.org/10.3389/fnagi.2016.00058
All Alzheimer's Products